Your basket is currently empty!
Instituto Gulbekian de Ciência researchers show how inheritance of Sickle Cell Anemia protects against Malaria infection
Instituto Gulbekian de Ciência researchers show how inheritance of Sickle Cell Anemia protects against Malaria infection
Sickle hemoglobin confers tolerance to Plasmodium infection.
Ferreira, A., et al. (2011). Sickle hemoglobin confers tolerance to Plasmodium infection. Cell. 145(3): 398-409. [PMID: 21529713]
| Malaria claims the life of a child every 45 seconds, but without the prevalence of sickle cell anemia in populations of sub-Saharan African descent, it would be even more lethal.
Sickle cell anemia is caused by a point mutation in the human hemoglobin (Hb) gene which leads to aggregation of Hb and characteristic “sickling” red cell morphology. While sickle cell disease is itself a serious and life-limiting condition, the sickle cell trait persists because inheritance of a single copy (each individual carries two) of mutant Hb is protective against malaria. |
 The Soares Lab (Ana Ferreira is in white, centre front) The Soares Lab (Ana Ferreira is in white, centre front) |
Since this survival advantage was first observed, its mechanism has remained elusive – a puzzle known to all high-school biology students.
Now, using ProImmune’s Pro5® MHC class I Pentamers for immune monitoring, a team at the Instituto Gulbenkian de Ciência in Portugal have shown exactly how sickle Hb (HbS) induces tolerance to malaria.
Malaria is caused by infection with Plasmodium parasites. In their study, Ferriera et alshow that the presence of HbS does not reduce parasite load, but instead impairs the development of malaria disease pathology via two distinct mechanisms.
The team established a mouse model for sickle cell disease and malaria: briefly, mice engineered to express human Hb or HbS were infected with an engineered variant of Plasmodium berghei, and scored for the development of experimental cerebral malaria (ECM). Their transgenic P. berghei was designed to express an MHC Class I epitope from herpes simplex virus glycoprotein B (SSIEFARL), and using ProImmune’s H-2Kb/ SSIEFARL Pro5® Pentamer, they were able to monitor pathogen-specific CD8+ T cell numbers accurately in experimental mice. The HbS mice fail to succumb to ECM after infection. Compared with wild-type mice, malaria-infected HbS mice showed decreased ECM pathology, including brain edema, and decreased brain accumulation of the CD8+ T cells implicated in ECM pathology (figure 1).
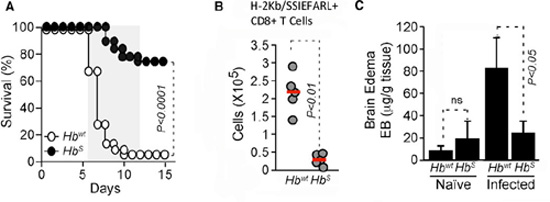
Figure 1. HbS Prevents ECM Onset
(A)Survival of Plasmodium infected Hbwt (n = 91) and HbS (n = 76) mice (10 independent experiments with survival advantage p < 0.05). Grey shading: expected time of ECM. HbS mice have a clear survival advantage. (B) Brain malaria-specific (H-2Kb/SSIEFARL Pentamer+ ) CD8+ T cell numbers infected Hbwt and HbS mice, 5 days after Plasmodium infection. Dots represent single mice (n = 4 -14/group). Red lines represent mean values. There are lower numbers of brain-infiltrating malaria-specific T cells in the HbS mice. (C) Mean brain edema in naive versus infected Hbwt and HbS mice ± standard deviation (n=4/group), 5 days after Plasmodium infection. Hbwt mice have significantly more edema than HbS mice. ns: not significant.
ECM pathology has previously been attributed to cytotoxic accumulation of free heme. Via a series of mouse crosses, Ferriera et al were able to establish how HbS counteracts heme accumulation. HbS induces expression of heme-oxygenase-1 (HO-1) in hematopietic cells. HO-1 –deficient mice, or mice in which HO-1 is pharmacologically inhibited, were no longer protected from ECM even when they expressed HbS. Mice deficient in the transcription factor Nrf2 were used to show that HO-1 expression relies upon Nrf2 expression. The team suggest that HO-1 exerts its protective effect via carbon monoxide (CO). CO is a by-product of heme catabolism by HO-1, and binds free Hb to inhibit further heme release.
Independently of its effects on HO-1, HbS affects the activation and expansion of malaria-specific CD8+ T cells. Using H-2Kb/ SSIEFARL Pentamer staining to compare Hb and HbS-expressing mice five days after infection, they found that there was no expansion of antigen-specific CD8+ T cells in the HbS mice, and that this lack of expansion was still evident when the infected mice were also HO-1-deficient. While this suggested that the T cell element of malaria pathology is independent of HO-1, there is still a role for heme itself in modulating T cell numbers. Administration of free heme to mice prior to infection was sufficient to limit epitope-specific T cell expansion.
The insights into malaria pathogenesis that Ferriera et al have gained using ProImmune Pentamers open up this longstanding mystery to further research efforts. Over the next few years we hope to see novel malaria treatments being developed to exploit this new knowledge of both the HO-1 dependent and the immunopathologic elements of malaria progression.
Figure reproduced with permission from Elsevier